
Research
I am a computational evolutionary biologist who focuses on developing methods that quantitatively analyze large genomic data to answer questions arising in biology, using techniques and concepts from statistics, mathematics, and computer science. The overarching theme of my research topics is to infer hybridization and phylogenetic networks using genomic data. My curiosity is not confined to a particular system but extends to all forms of life.
Inferring phylogenetic networks from the sequence data using composite likelihood
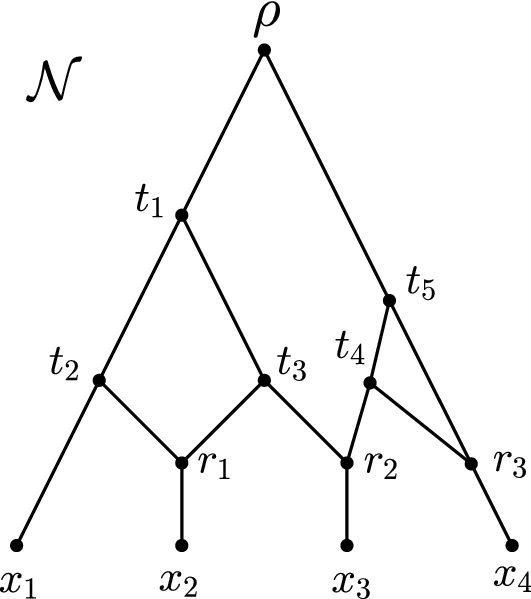
While phylogenetic networks (see right) can model more complex biological histories that the bifurcating trees cannot, the use of networks in evolutionary investigations is limited by the inability of existing methods to handle large datasets due to their algorithmic complexity. I propose a novel method, PhyNEST (Phylogenetic Network Estimation using SiTe Patterns), for inferring phylogenetic networks directly from sequence data. PhyNEST achieves computational efficiency by employing the composite likelihood framework in the network setting and accuracy by incorporating all sources of variability in genome data through quartet site pattern frequencies. Composite likelihood approximates the likelihood; however, it has been shown to be extremely useful in phylogenetic context and may offer a promising direction to alleviate the computational burden required to compute the full likelihood. The method is implemented in publicly available, open-source software [download at github].
Related publications
- Phylogenetic networks empower biodiversity research
- Inference of phylogenetic networks from sequence data using composite likelihood
- Classes of explicit phylogenetic networks and their biological and mathematical significance
- Digest: Frequent hybridization in Darevskia rarely leads to the evolution of asexuality
Detecting hybridization using genomic data
The surge in genome-scale data has revolutionized hybridization studies, shifting focus from merely detecting the presence or absence of hybrids in nature to investigating their genomic constitution and evolutionary dynamics. Despite several statistically sounding methods are available, accurate detection of hybridization from genomic data remains a challenging task, particularly under certain biological scenarios (e.g., ancient and/or asymmetric hybridization and high amount of incomplete lineage sorting). Using simulation, I have presented that site-pattern-frequency-based methods are more reliable in detecting hybridization and estimating relevant parameters than population clustering methods in various biological scenarios. Through collaboration with biologists, I am investigating evolutionary histories in biological systems suspected of hybridization. However, I argue in that results from hybrid detection methods should be cautiously interpreted, as they can be sometimes unreliable, for instance, in the presence of parthenogenetic hybrids. Recently, I contributed in developing a novel hybrid detection method using likelihood ratio test.
Related publications
- A Likelihood ratio test for hybridization under the multispecies coalescent
- Comparative performance of popular methods for hybrid detection using genomic data
- Anchored hybrid enrichment resolves the Lacunicambarus (Decapoda: Cambaridae) phylogeny
- Digest: Frequent hybridization in Darevskia rarely leads to the evolution of asexuality
- There and back again: The unexpected journeys of Metridium de Blainville, 1824 between the old oceans and throughout the modern world
- A likelihood ratio test for hybridization under the multispecies coalescent
Theoretical evaluation of abstract networks in evolutionary investigations
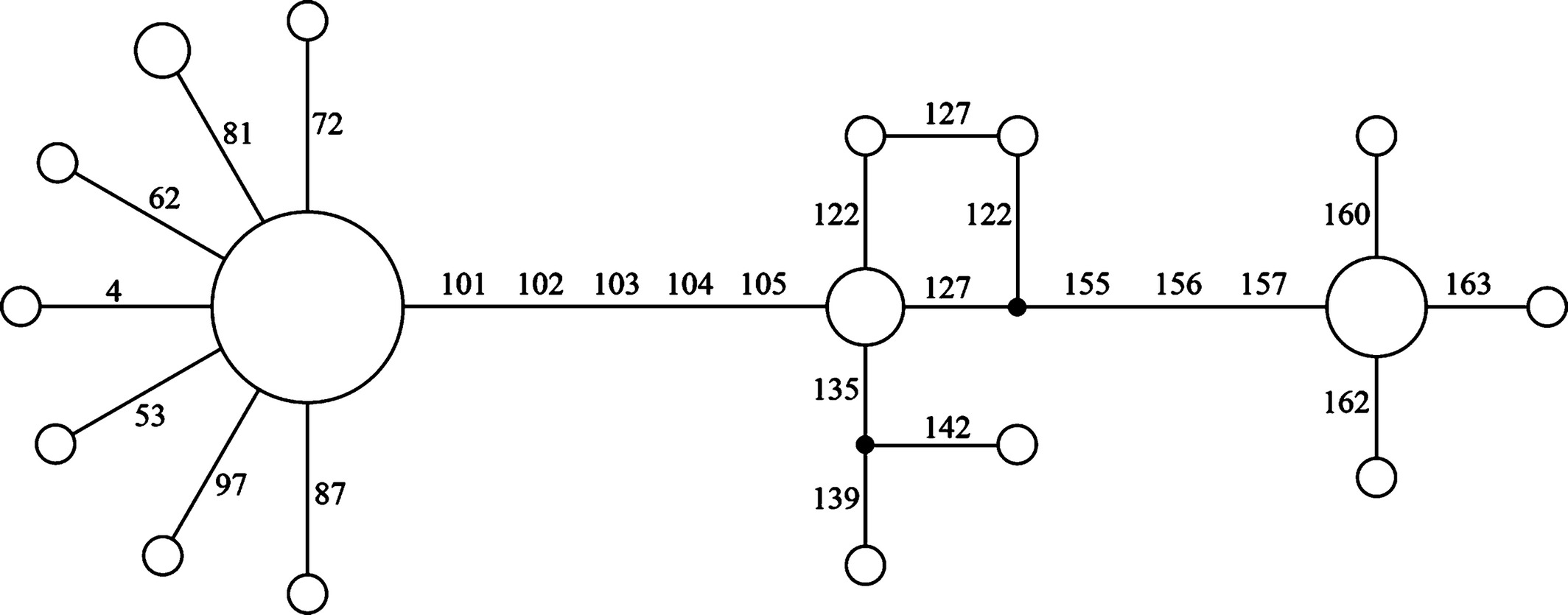
Two types of networks are being used in practice: abstract and explicit networks. It is crucial to distinguish between abstract networks, drawn to depict non-tree-like signals in the data without modeling the causes of this deviation, and approaches modeling violations of tree-like evolution due to biological processes like hybridization. I focus on the popular abstract network method that computes Median-Joining Networks (MJNs), which is primarily designed to visualize patterns and conflicting signals in DNA data. I have philosophically criticized the use of MJNs in evolutionary investigations due to inherent unrealistic assumptions and algorithmic limitations. I have criticized the inappropriate use and over-interpretation of MJN in understanding the evolution of SARS-CoV-2. Moreover, I analyzed 85 published datasets, revealing significant differences between relationships computed in MJN and those estimated in Bayesian phylogeny in over one-third of cases. This conclusion reinforces the decades-old recognition that phenetic-based algorithms are not a defensible way to hypothesize evolutionary relationships. Despite its inappropriateness, MJN remains popular, leading to indiscriminate usage in various applications.